

 Calling all SWATH® Acquisition users!
Calling all SWATH® Acquisition users!
If you have a TripleTOF® System and you’re using SWATH Acquisition for your quantitative proteomics experiments, you’ve made the right choice. SWATH is a data independent workflow that enables data to be acquired for every detectable analyte in a complex sample. You create a digital map of the sample that can be mined for new information any time new biological insights are hypothesized. It’s all there!
Since the introduction of the workflow at HUPO 2010 in Sydney, Australia, we have been working to further improve the workflow. Optimizations in data acquisition such as smaller and variable Q1 window widths and increased instrument dynamic range have provided increased depth of coverage for your proteomics sample, without compromising reproducibility or data quality.
But what about data processing? Can we improve even further?
The answer is yes! Typically an ion library is used for targeted processing of SWATH data for peptide and protein identification and quantitation. The ion library contains the masses of the peptide ions, sequence-specific fragment ions, relative fragment intensities, and relative retention times, and is easily generated by performing a simple data-dependent acquisition and database search.
In this study, scientists from SCIEX demonstrate that for biological systems that will be studied repeatedly, it is worth taking the extra time to create a deeper ion library as much more information can be extracted from the SWATH acquisition data.
For example, SWATH replicates of a HEK human cell lysate were acquired and processed using three different libraries:
- A simple 1D human cell line library generated using traditional data-dependent acquisition strategy (IDA)
- A 2D human cell line library using a more in-depth 2D off-line fractionation/IDA strategy
- A Pan-Human library (PHL) library using a large number of different human cell lines extensive fractionation and IDA strategy
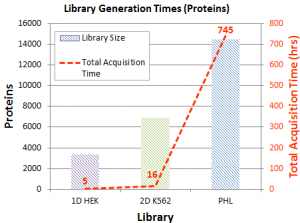
Figure 1. The Impact of Deeper Ion Libraries on Extraction of Quantitative Data from Human Cell Lysate SWATH® Acquisition Data.
As shown in Figure 1, a 118% gain in quantified proteins was observed using a simple ion library (1D HEK) and an extensive ion library (PHL). And the quality and reproducibility of the quantitation are maintained even into the low abundant protein/peptide regime.
A researcher can balance the library generation time with the depth of coverage needed. The three libraries outlined here took increasingly more time to generate. A simple IDA experiment can take only a matter of hours. A 2D fractionation followed by LC-MS/MS on each fraction can be performed in a matter of days. And some groups have invested significantly more time in library generation using multiple cell types and a large degree of fractionation to cover as much of the proteome as possible.
This work also highlights the superior dynamic range of quantitative information that is present in a SWATH Acquisition data file over the traditional data dependent approaches for quantitative proteomics. To read more details of this work, and to see what size
To read more details of this work, and to see what size ion library would make the most sense for your biological system of study, download the full technical note.



 Contact Support
Contact Support
0 Comments