
The “Intensity (Peptide)” values come from LCMSReconstruct, in ProteinPilot software 5.0. It maps the RT, m/z, intensity MS1 surface to find the peak information for the peptide. The Intensity (Peptide) is a weighted sum of the heights of the isotope series at the determined apex of the peptide elution, after doing a 3-point moving average smoothing (in both m/z and time). The weighted sum is based on the expected pattern of the isotopes, where the weight is reflective of the significance of the isotope. If the pattern is close to expected, it will be the simple sum, but if it deviates from expected, then it will be somewhat less than the sum (due to e.g. convolved peaks, distortion, interferences, etc).
As you can see, because of the nature of the underlying algorithm (the moving average smooth and the proprietary charge filtering), you will not be able to reproduce the Peptide Intensity found in the ProteinPilot export using the processing algorithms available in PeakView software. Note that the manual smoothing options in PeakView are different as well (option under AutoProcessing).
Note there is also a PrecursorIntensityAcquisition column in the exports, this is computed at the point of the MS/MS acquisition (which is not necessarily at the peak apex). This is computed in a similar way as above – it is a charge filtered value that is derived from interpolation (since the MS1 scan and MS/MS scan do not occur at the same exact same point in time).
In the development of the LCMSReconstruct algorithm, we found that using the weighted sum of the heights of the isotope series was superior in quantitation testing to using the extracted ion chromatogram (XIC) area or the feature volume approaches for survey level relative quantitation applications. It was also found that the LCMSReconstruct signal processing method also improved mass accuracy, yielded increased protein, peptide, and spectra identification by up to 10%, and reduced the number of incorrect monoisotopic mass determinations by several-fold (HUPO 2012 Poster 092 – Shilov, et al.).
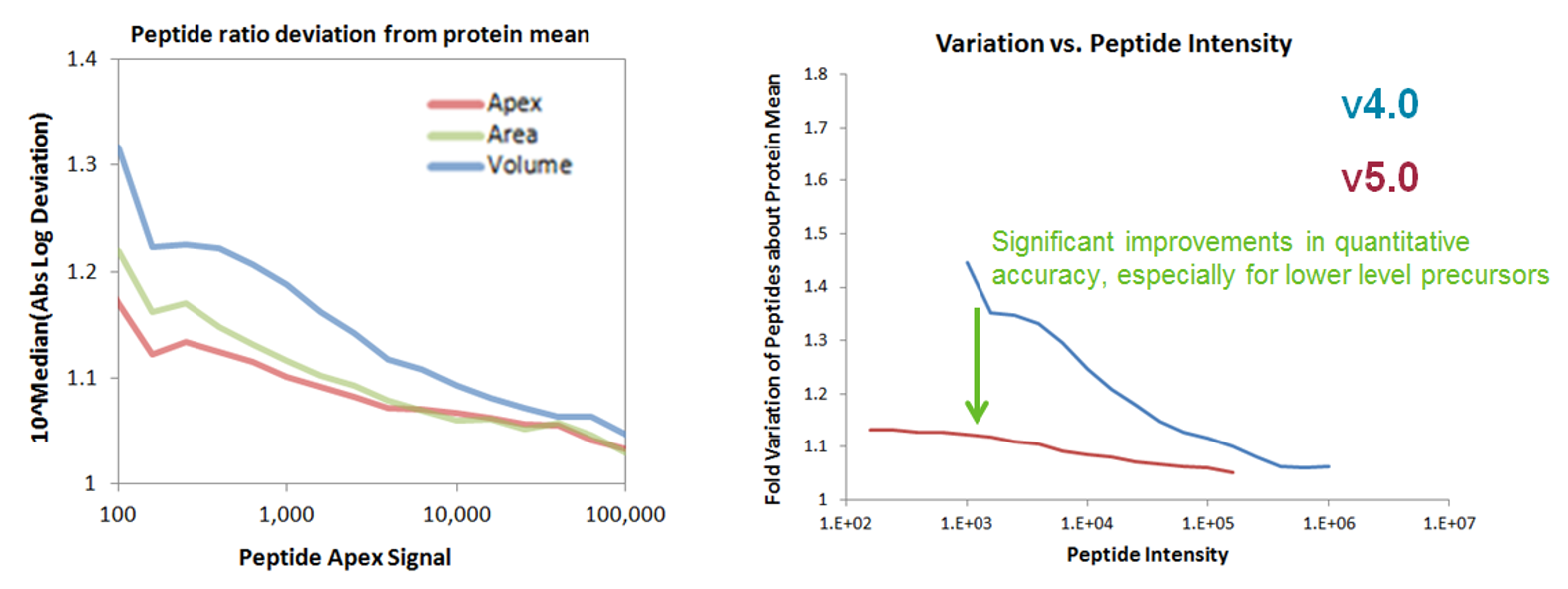
Above left: Peptide intensity ratio at the elution apex shows the smallest variance as it relates to the protein-specific ratios (mean of all peptides per protein).
Above right: Compared to previous versions, ProteinPilot 5.0 using LCMSReconstruct reduces the variance of peptide intensity ratio, as it relates to the protein-specific ratio, especially for peptides with lower intensity.
0 Comments