
In an LC-MS experiment there are multiple sources of variance that can confound the quality of your results. This variation can be biological e.g. differences between treated and control groups, but can also be non-biological, usually from small variations in experimental conditions.1 These variations can include differences in sample preparation and handling (e.g. different starting amount of protein), or differences in instrument performance (e.g. variance across batches).
The goal of normalization is to make the sample data more comparable, and the downstream analysis more reliable by bringing a set of values across multiple samples onto a common scale. In the case of mass spectrometry data, this means bringing the measured MS features onto the same area scale so that we can compare across samples with reduced variance.
There are many different normalization methods in the literature (see Välikangas et al. 2018 for a good review)2, and within MarkerView software. The correct choice depends on the experimental design and data type. Here, we discuss the recommended normalization options. More information on all of the normalization options is included in the MarkerView software Reference Guide that installs in the Help menu of the software.
Normalize LC-MS using internal standards – specific features within the dataset are defined as internal standards, and used to calculate a normalization factor for each sample. The normalization factor is calculated as:

Finally, a new table of areas is computed that has been corrected by the normalization factor. If multiple internal standards are used, the final normalization factor for each sample is an average of all available internal standards for that sample. This normalization method is appropriate for most accurate work when internal standard(s) have been added to all samples.
Normalize using MLR method – the MLR (most likely ratio) algorithm is new in MarkerView software 1.3 and is more robust for typical MS data than some other normalization techniques. This algorithm works best for normally distributed data where it can be assumed that the bulk of analytes are not changing. This is the best normalization option to use for most MS data (when internal standards are not used) based on our testing. The only limitation for this approach is that the dataset must possess enough variables where most are not changing significantly.
MLR can perform multiple levels of normalization starting with technical replicates (that is, samples with the most similarity) followed by normalization across experimental groups. Note that this multi-level support is available in MarkerView Software 1.3.1). MLR can be run on any MarkerView Software table, at the fragment or analyte level, or at the fragment/peptide or protein level.
To define the hierarchical metadata that MLR can use, enter the group information as ExperimentalGroup.BiologicalReplicate. Grouping is from left to right, separated by a period; samples with the same group name are assumed to be technical replicates.
Normalize using total area sums – the total response (sum of all the peak areas) is computed and then the scale factor is computed for each sample as:
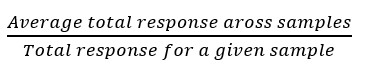
This technique is recommended when the majority of features in the dataset are expected to be unchanged, for example, when profiling samples of similar complexity, like in omics experiments. This strategy is sometimes used but, typically, we would recommend MLR normalization over total area sums.
Normalize using manual scale factors – with this option, you can directly enter specific normalization factors to each sample that were determined by other means. Some examples of manual scale factors include sample volume, amount of loaded protein or sampling rate.
Helpful references:
- Karpievitch YV, Dabney AR, Smith RD. 2012. Normalization and missing value imputation for label-free LC-MS analysis. BMC Bioinformatics. 13: S5.
RUO-MKT-18-12137-A
0 Comments